🧬
Stalling probability calculation
Computes residue-level stalling probabilities using local A-site features and weighted neighbourhood context, with optional motif-folder support for machine-learning-derived pausing maps.
StaulliQ is a research software platform for sequence-resolved analysis of ribosomal stalling, enhanced Chou–Fasman secondary-structure prediction, AlphaFold/DSSP comparison, chemistry-aware clustering, and batch-scale exploration of co-translational folding.

Developed in the Vincent Kelly lab within the Trinity biochemistry / biochemical sciences research environment, with focus on queuine-linked translational control and nascent-chain folding.
The platform was built to move beyond simple pause-site detection and instead explain why stalling occurs and how it connects to secondary-structure emergence in the ribosome exit tunnel. It integrates a stalling predictor with an enhanced Chou–Fasman framework and structure comparison tools in one data-driven workflow.
Quantifies electrostatic charge, hydrophobicity, polarity, aromaticity, proline content, and queuine-linked effects across an upstream tunnel-sized window.
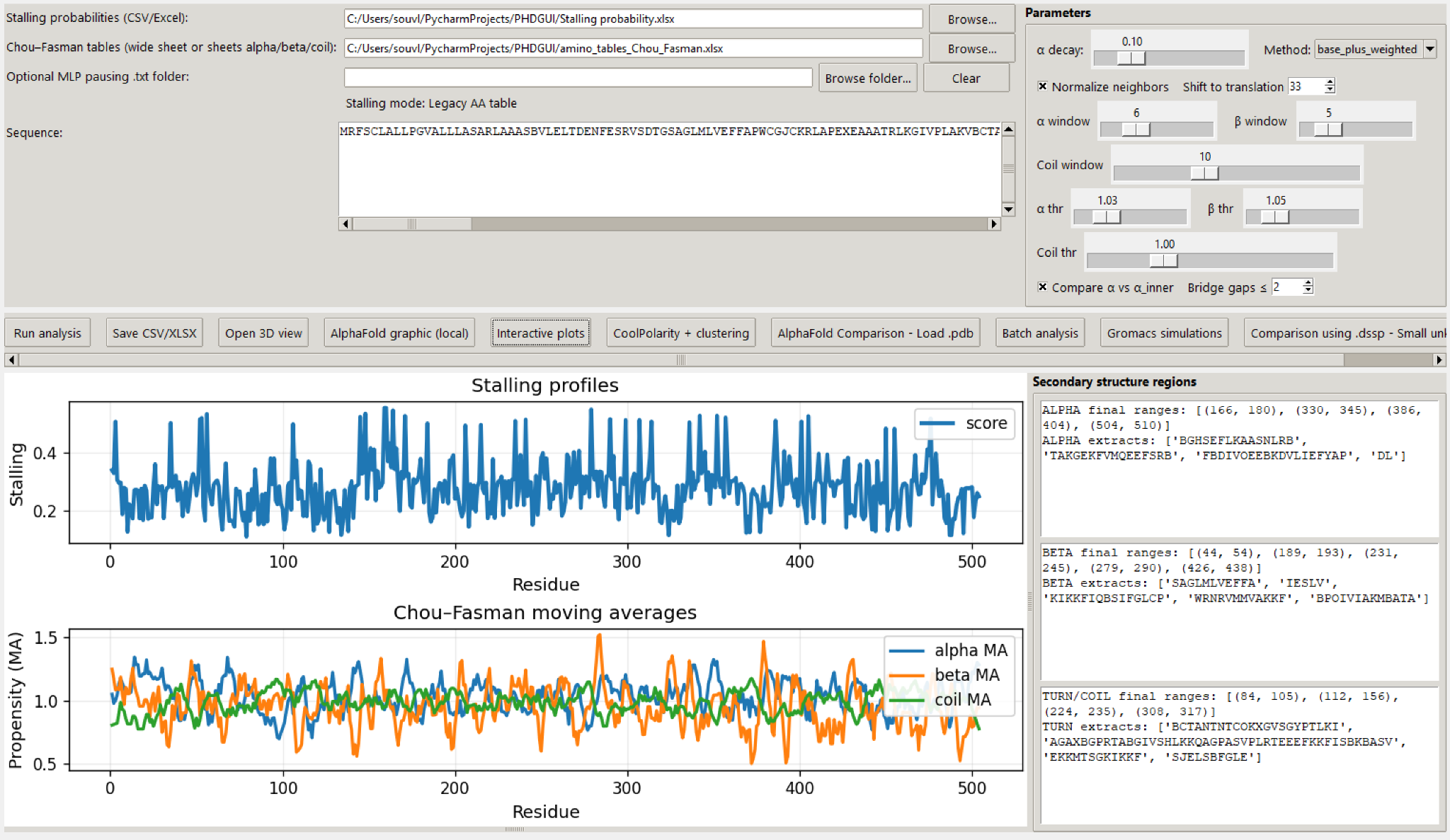
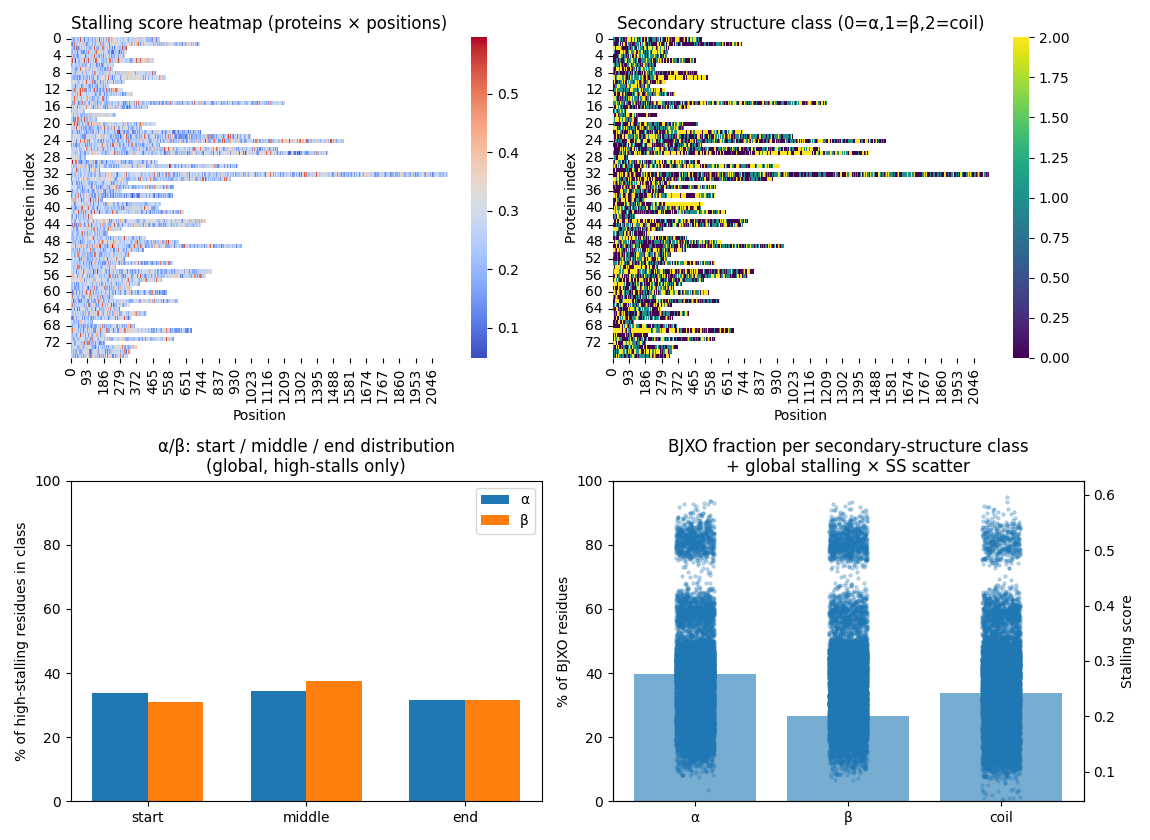
Maps A-site stalling to α-helix, β-strand, and coil formation at the ribosome exit using a tunable Chou–Fasman implementation.
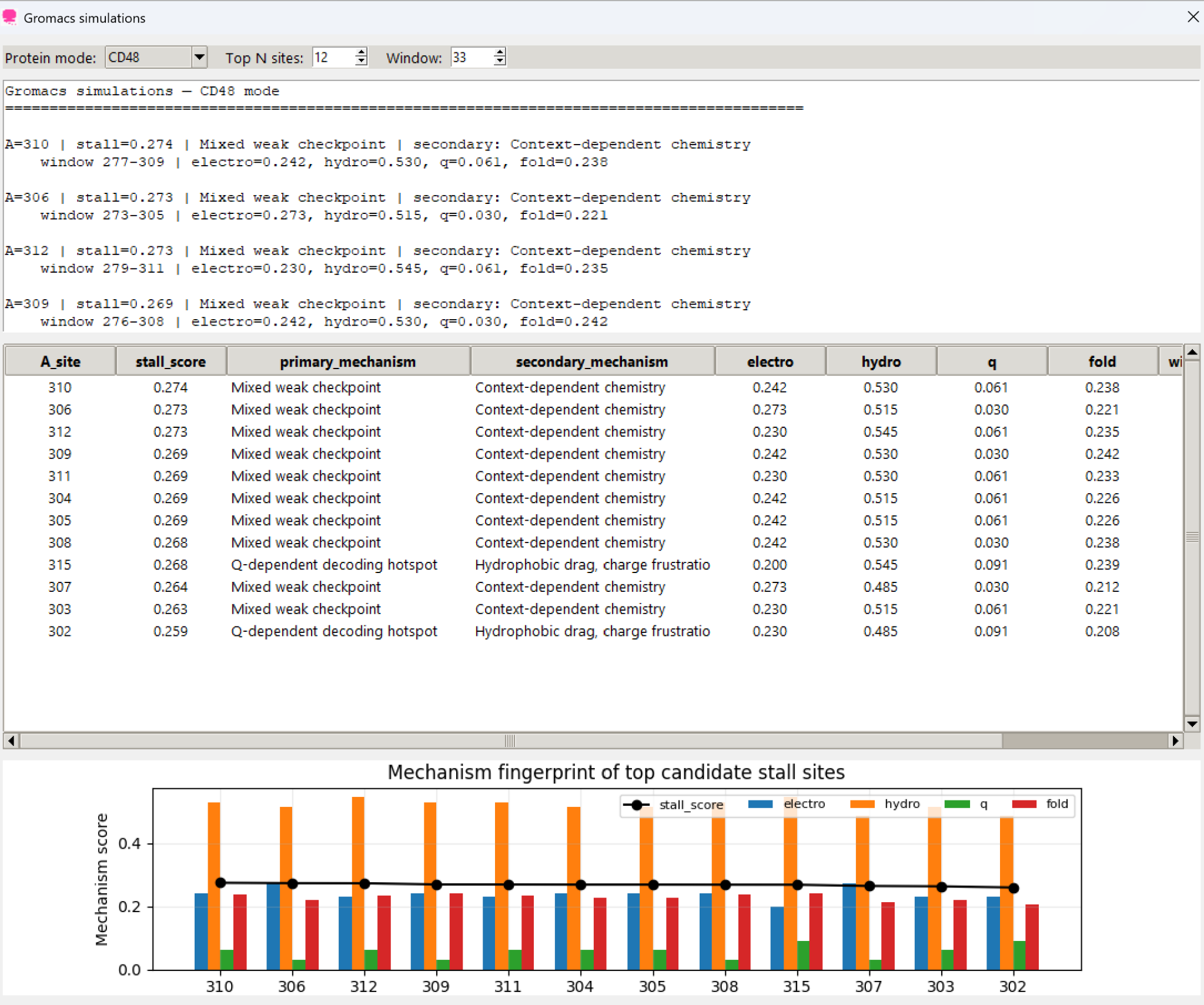
Separates likely causes of slowdown into electrostatic braking, hydrophobic friction, folding checkpoints, and Q-dependent decoding modes.
StaulliQ is presented here as a research-facing software page for Trinity College Dublin. The site highlights the software identity, interface, and analytical modules while providing a clear route for installation and lab attribution.
The current interface exposes separate analysis paths for single-sequence prediction, optional motif-based pausing input, structural comparison, chemistry-aware clustering, 3D viewing, and batch workflows. The uploaded Python GUI also includes dedicated windows for AlphaFold comparison, CoolPolarity clustering, batch protein inspection, and Gromacs simulations.
Computes residue-level stalling probabilities using local A-site features and weighted neighbourhood context, with optional motif-folder support for machine-learning-derived pausing maps.
Predicts α, β, and coil states with tunable windows, thresholds, and support for modified residue propensities under ribosome-constrained conditions.
Decomposes slowdown into electrostatic braking, hydrophobic friction, folding checkpoints, and queuine-dependent decoding hotspots.
Groups high-stalling residues by local chemistry context to identify positive-biased and hydrophobic-enriched environments along the sequence.
Compares Chou–Fasman-derived structure assignments against uploaded .pdb or .dssp data to highlight agreement, mismatches, and confidence patterns.
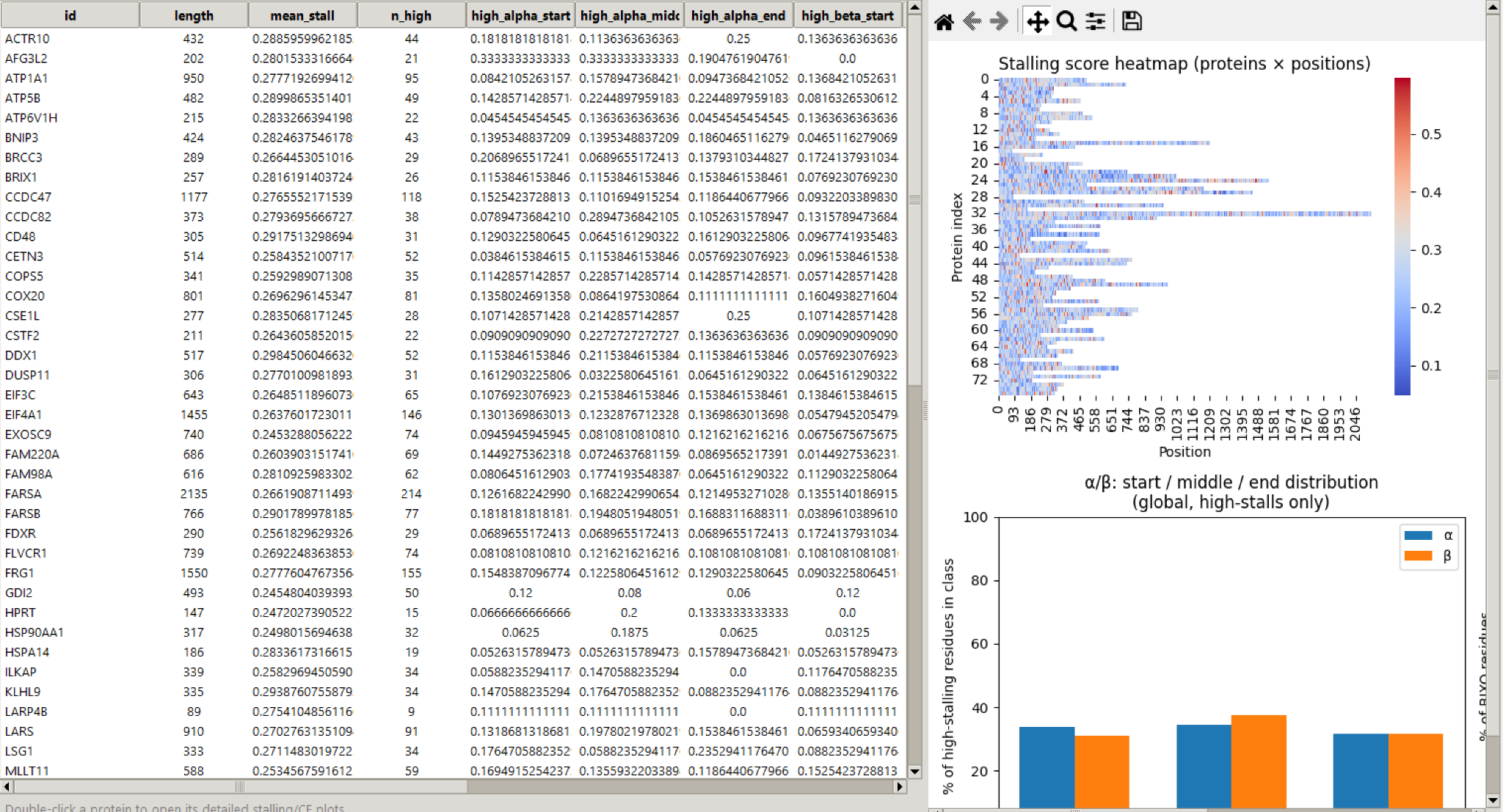
Processes multiple proteins at once, generates heatmaps and regional summaries, and opens per-protein inspector views for targeted follow-up.
The gallery below is built from the interface and analysis images you provided. It gives visitors a quick visual path through StaulliQ’s major modules, from single-protein scoring to chemistry clustering and batch analytics.
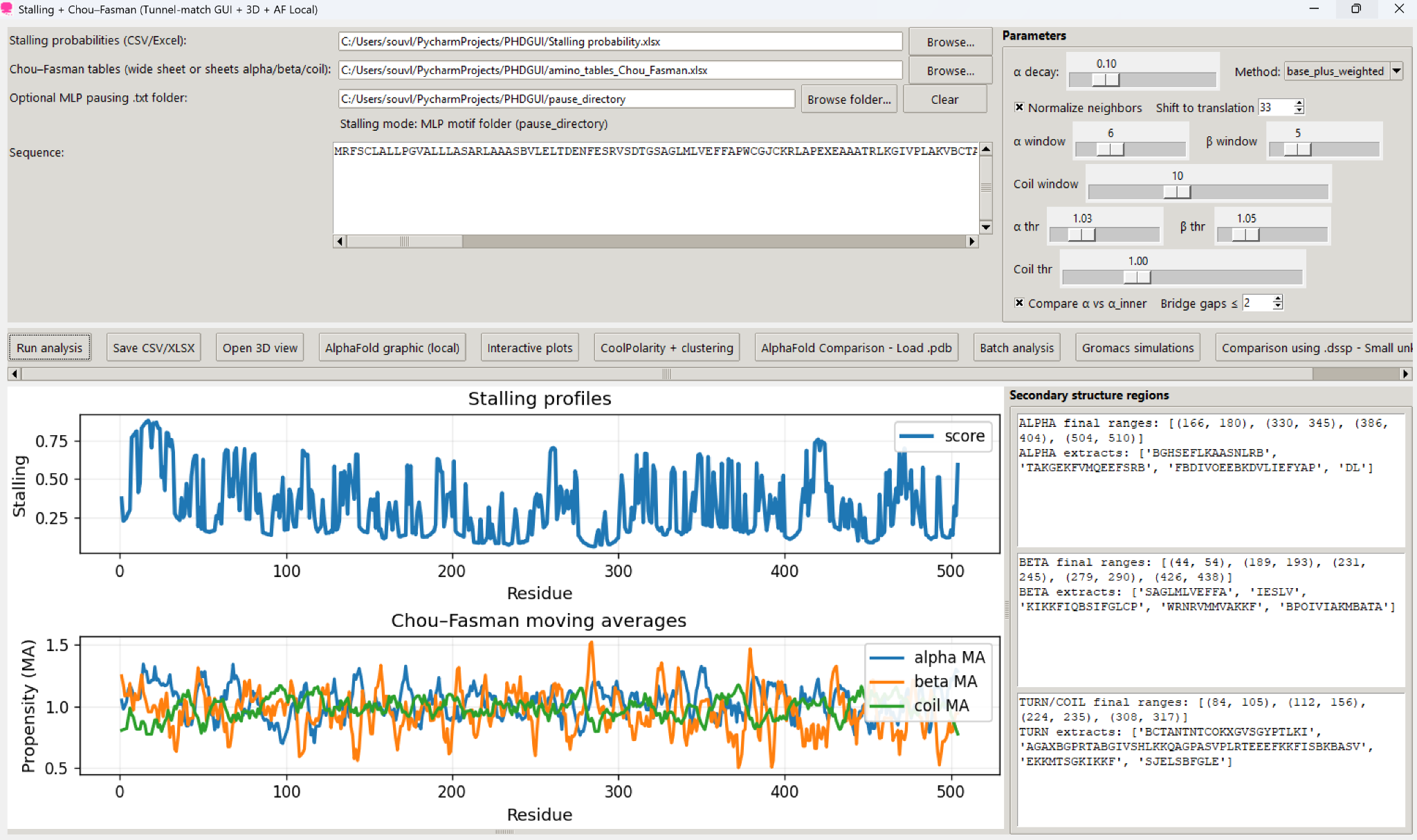
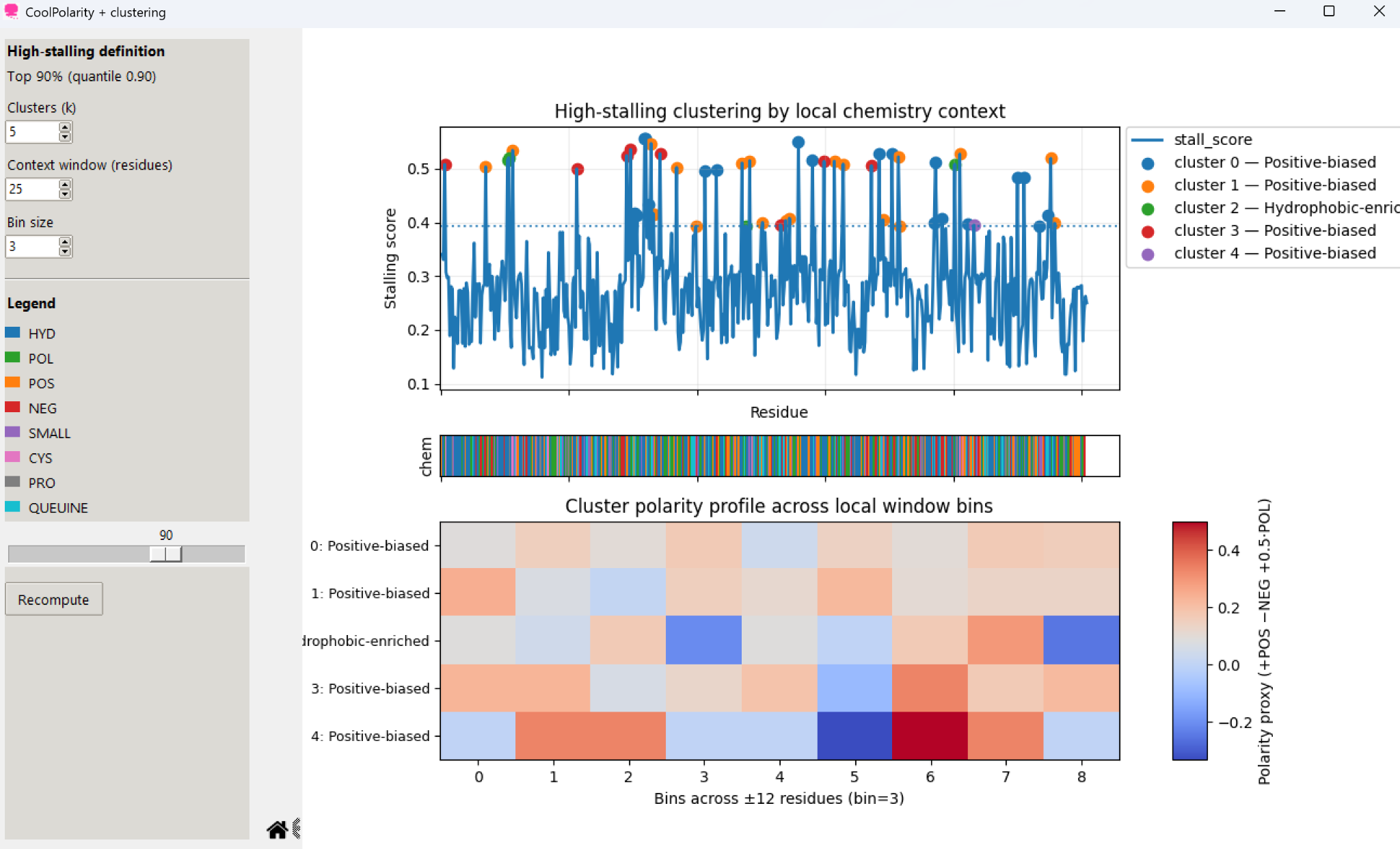
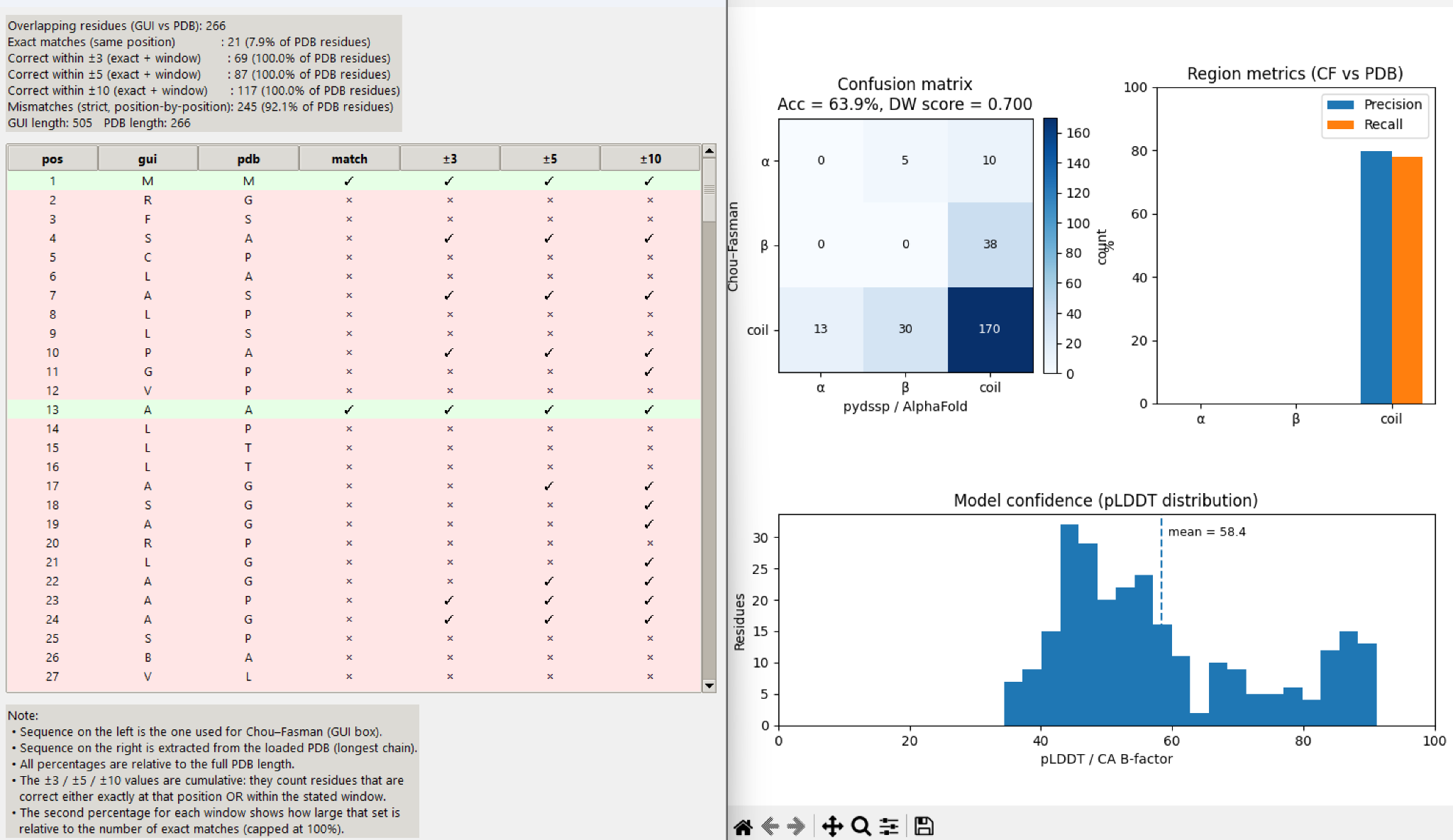
StaulliQ is organised as a modular analysis environment so users can move from raw sequence input to interpretation without leaving the interface.
Bring in stalling tables, Chou–Fasman parameter sheets, optional pausing folders, and gene or protein sequences.
Generate per-residue stalling scores using weighted neighbourhood context and optional motif-derived probabilities.
Overlay Chou–Fasman predictions, compare with AlphaFold/DSSP outputs, and inspect residues emerging at the ribosome exit.
Save CSV/XLSX outputs, screen multiple proteins, inspect top candidate sites, and communicate findings with linked figures.
The current downloadable package contains the example input files and sample pause directory needed to run the software once the .exe is placed into the release bundle.
Stalling probability.xlsxamino_tables_Chou_Fasman.xlsxpause_directory/ with all .txt pausing filesfull_gene_list_short.xlsxThe website build already points to the sample package. You can later replace or extend this zip with the final Windows executable and keep the same download button.
Open the instructions page for a short guided setup: launch the .exe, load the stalling file and amino-acid table, confirm the default PDIA3 sequence, and press Run analysis.